AD is the most common neurodegenerative cause of dementia in the elderly, affecting ~6% of the global population aged >65 years. AD causes neuropsychiatric symptoms, emotional disturbance, and the progressive impairment of regular activity, resulting in dependence, disability, and mortality.
Both the familial and the sporadic forms of AD share a typical phenotype with at least eight features:
(i) the progressive accumulation of senile plaques containing the protein fragment β-amyloid (Aβ) and neurofibrillary tangles (NFTs) containing hyperphosphorylated tau protein;
(ii) synaptic signaling deficits;
(iii) neurite and brain cell atrophy;
(iv) uncontrolled oxidative stress;
(v) enhanced pro-inflammatory signaling;
(vi) changes in innate immune signaling;
(vii) progressive changes in gene expression differing from healthy brain aging;
(vii) progressive cognitive impairment and dementia.
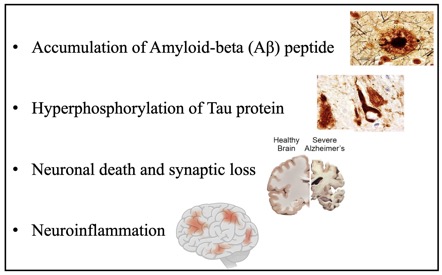
Due to ineffective therapy, there is currently neither a cure nor adequate clinical treatment for AD. How AD originates and propagates through the brain and central nervous system (CNS) remains unclear. Brain aging, especially during AD, is associated with increased activation of innate immune pathways and an increased activation state of microglia and astrocytes, frequently referred to as neuroinflammation. Constant activation of these cells potentiates neuronal damage, provokes alteration of the blood-brain barrier (BBB), and disruption of the blood vessels permeability, thus facilitating brain infiltration of the immune cells. Copious evidence from clinical and experimental research, including our own, suggests that T lymphocytes and neutrophils migrate into the AD brain. In particular, the biological functions exerted by neutrophils in Alzheimer’s are somewhat more extensive than previously thought.
The new concept emerging from the recent literature states that AD should be considered not just as a neurological but a systemic disease; inflammation is not restricted to neuroinflammation but also encompasses systemic inflammation with a sustained interaction between the peripheral, systemic, and CNS inflammatory compartments. Therefore, perturbation at any level of this network will propagate dysregulation throughout the entire circuit. Considering AD as a SYSTEMIC IMMUNE PROCESS raises important questions about how communication between the peripheral and central compartments occurs and whether this crosstalk represents a therapeutic target.
We perform our studies using mainly two TRANSGENIC AD-like disease mice: 5xFAD and 3xTg-AD.

Recently, we started exploring inflammatory changes occurring in SPORADIC vascular AD mice (polyI:C). The PolyI:C mice display low-grade inflammation over their lifetime, which spreads to the brain, causing an AD-like phenotype with amyloid and tau proteinopathy, neuroinflammation, vascular remodeling, and upregulation of vascular biomarkers.